We classify each lasso structure according to two different criterion:
described below.The piercing information (described in Lasso detection section) obtained for each closed loop in a chain enables us to classify the covalent loops, as well as the whole chains. Currently, based on survey performed in [1-2] we distinguish five major classes of pierced loops and one class containing unpierced loops (Fig. 1):

Fig. 1 Schematic depiction of major classes of lasso typesfound in the Protein Data Bank. First row from left to right L0, L1, L2, L3, and second row LS (left) and LL (right).
Each major class can be however split to subclasses depending on the direction of crossing and the tail, which pierces the surfaces (N- or C-terminal) as described in Spanning a surface on the loop (Fig. 3 and 4). For example the L1 class splits into L+1N, L-1N, L+1C and L-1C, (Fig. 2) where the sign describes the direction of crossing, and the letter the crossing tail.
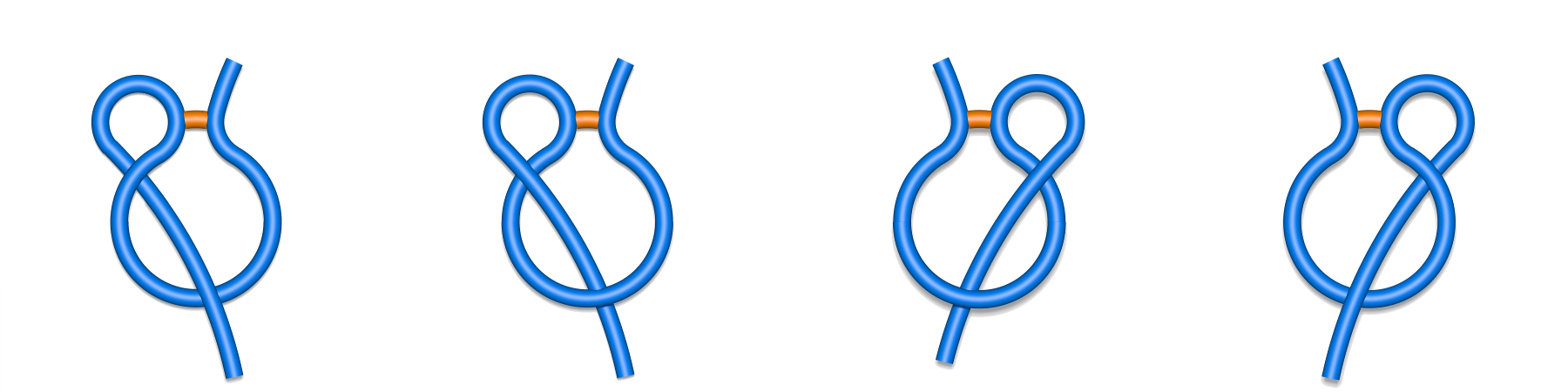
Fig. 2 Schematic depiction of subclasses of single lasso class L1: L-1N, L+1N, L+1C, L-1C (class are shown respectively from left to right). The N-terminal tail is placed in the left side of the picture, the C-terminal in the right side.
The same splitting occurs in the other classes giving rise to 28 subclasses in total. In the case of double/triple lasso, as the piericngs are alternating, it suffices to write only the sign of sequentially first crossing (Fig. 3). On the other hand, the chains with supercoiling are described by a full list of "+" and "-" signs.

Fig. 3 Schematic depiction of two L2 subclassess: L+2C (left) and L-2N (right).
There are however some proteins possessing more than one covalent loop pierced. These should be distinguished from the proteins with only one closed loop threaded. Therefore, we can classify whole protein chains according to the lasso motif of their closed loops. To this end we print for every protein chain the topological classes of its covalent loops in sequential order of the loop opening residues, separated by a whitespace (see Fig. 4). We however suppress L0 loops, as they are trivial. Moreover we join the same classes, i.e. we write 2L2 instead of L2 L2. Here, to maintain the clarity of the overall view, we do not split the classes into subclasses. This should be stressed, because such notation, although simple, is not universal. There can be potentially topologically different proteins with the same class prescribed.
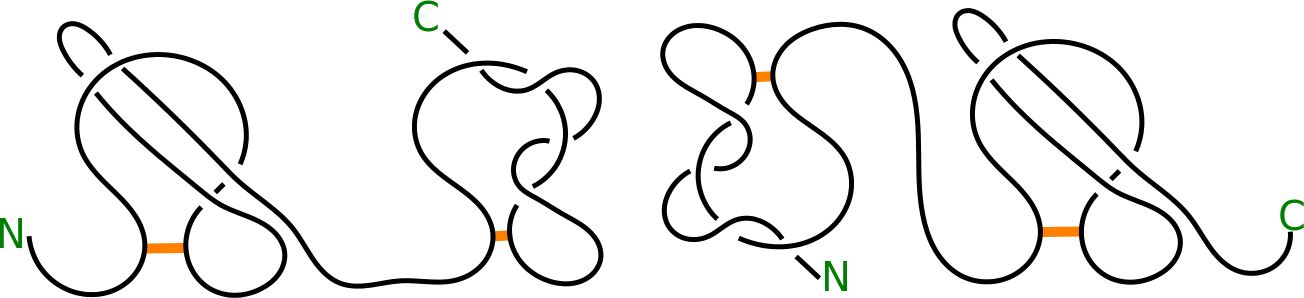
Fig. 4 Schematic depiction of proteins with L2 LS (left panel) and LS L2 (right panel) topology.
Remark: it should be noted that in general lasso could be more complex. To take this into account the server allows the detection of any type of complex lasso, however the automatic name will be given only to the five class mentioned above.
The most common loop closing bridge is the disulphide bridge. However, this is not the only possible way of closed loop formation. In particular the side groups of amino acids can be naturally joined via C-N, C-O or C-S bond (in amide, ester or thioester for example). Therefore LassoProt distinguishes the loops into the following short-named categories: SS-bridge (structures with cysteine bridge); Amide (joined by C-N interaction); Ester (joined by C-O interaction); Thioester (joined by C-S interaction) and other. Details about all the categories are given below. We would like to stress the fact, that only cysteine bridge is formed with ease in natural conditions. Other loop closing bonds are formed in posttranslational enzymatic, or autocatalytic reaction, or are introduced artificially.
SS-bridge
This category includes covalent loops with cysteine, or a disulfide bridge, which is the most abundant modification joining two side groups in proteins. It is formed in oxidizing conditions upon dimerization of free thiol groups of two cysteines with creation of S-S bond. The disulfide linkage in natural condition can also be formed by the action of specialized enzymes. In the pdb file it is identified by a line starting with “SSBOND” string, followed by indices of residues forming the bridge. Sometimes however, some cysteine bonds are contained in lines starting with "LINK", as joining Sγ atoms of cyteine residues. These are also treated as a regular cysteine bond. As the existence of this bond depends on the oxidizing potential of the solution (in reducing condition the S-S bond is torn apart with recreation of free -SH groups), both forms of proteins (oxidized with closed loop and reduced without it) can be found in Protein Data Bank. Therefore there can be two identical (according to sequence) structures, differing in topology only.
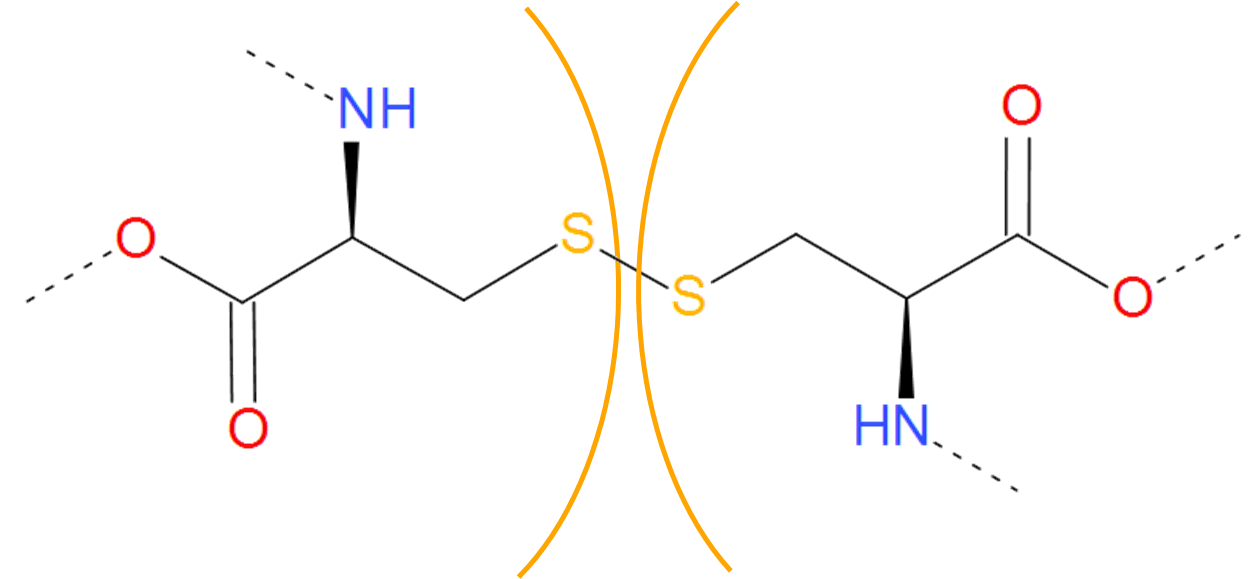
Fig. 5 Two cysteines forming cysteine bridge. The cysteines are separated by orange arcs. Dashed lines denote the remaining part of the protein chain.
The disulfide bond in proteins has at least two possible functions. One is the redox sensor relying on the formation/breaking of the bond in oxidizing/reducing conditions. The second role is holding the chain together reducing its freedom of movement, which enhances stability of the protein. This function is observed mainly in the secreted proteins, as in the reducing conditions present in cell compartments the cysteine bridges are unstable.
To maintain the rigidity-based stability of the protein chain in cells the other types of bridges can be useful. The information about other bonding types are found in the "LINK" line in pdb file. These lines contain mostly data of non-standard connection found in protein chains. This information include bonding atom types and indices of residues connected. However, these lines can store the information about protein - metal ion or protein - glycoside connection. Therefore, in searching for intrachain bridges, first we filter the information in these lines to extract only those bonds, which join amino acids within the same chain. These are then divided into four groups based on the atoms forming the bond. However, one has to be very careful, when analyzing the data based on the information in LINK line. This information is generated automatically, based on the distances within the structure. If the structure is wrong, the information if LINK line can be false.
Amide
The amide category is a short name for all bridges closed by the connection via C-N bond. Apart from most common amide bridges it also contains the amine structures formed by joining two residues. In most cases, the N-C bond is introduced posttranslationally (or aritificially) by the action of specified enzymes. However, autocatalytic mechanism is also possible (as during GFP functional unit formation). An amide bridge arises upon joining free amine and carboxylic groups. The free amine group can be found in side group of lysine and on N-terminus of the protein. The free carboxylic group can be found in the side group of aspartic and glutamic acids as well as on C-terminus of protein. Therefore, the amide linkage occures e.g. when one of the termini is condensed with the interior of the chain, or the termini are joined together (in cyclotide proteins). In the latter case however, one cannot distinguish the termini properly without the "source code" of RNA or DNA which was translated to this protein. From the viewpoint of LassoProt database, the most interesting proteines/peptides with such bridge are e.g. miniproteins with L1 topology introduced by the existence of the amide-based loop.
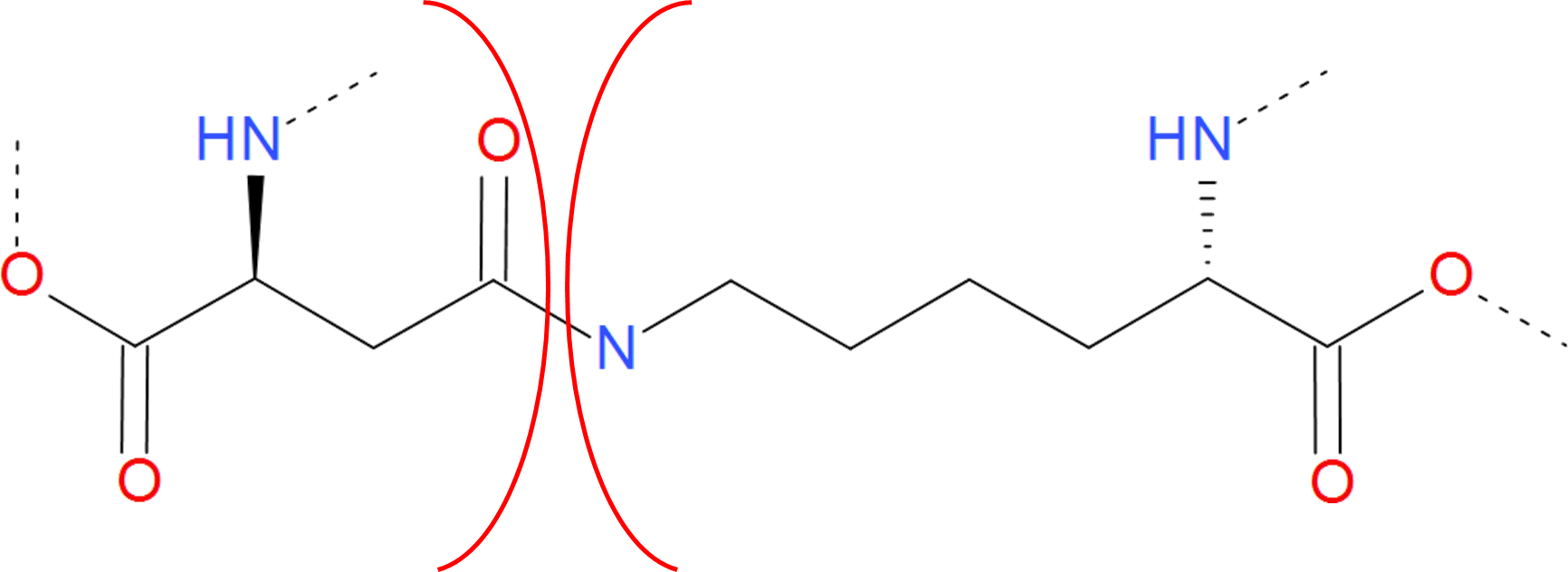
Fig. 6 Example of amide bridge between aspartic acid and lysine. The residues are separated by red arcs. The dashed lines denote the rest of the protein chain.
The amide bond can be viewed as the peptide bond, present not in the main chain. Therefore, in proteins it is often called isopeptide bond. Although amide bond is not prone to degradation in reducing/oxidizing conditions, it breaks in low or high pH. The amide bond due to theresonance effect is considered to be partially double. As a result it is stronger and shorter than the e.g. cysteine bond. Moreover, there is no freedom of rotation around the bond axis. The “amine bond” is also formed by joining a carbon and a nitrogen atom. This however, has no keto oxygen (oxygen atom joined by double bond to carbon atom). As a result the electron structure of the bond changes. The bond is single and there is no resonance structure impeding rotation around it. The free electron pair of nitrogen atom is not involved in any resonance structure, therefore the nitrogen atom is still basic (on the contrary, amids are not basic). The bond is not easily broken during hydrolysis. It is therefore more stable, than the amide bond. Similarly, it is formed naturally by the action of specialized enzymes, or autocatalitically. However, as such interactions are in most cases local, the loops introduced by amine bond are often too small and are not taken into our analysis.

Fig. 7 One of the "amine" bond existing in proteins. Tyrosine is joined with histidine. The residues are separated by the red arcs. The dashed line denote the rest remaining part of the protein.
In both cases however, one has to be careful and check, if the introduction of LINK as denoted in the pdb file actually introduces a loop. It can happen, that the amide/amine linkage is formed during a non-standard substitution of one of the terminus by some amino acid with formation of C-N bond. Moreover, the LINK method of joining N and C atoms is common in the description of non-standard amino acids incorporated into the chain, instead of a conventional bond. In both cases the closed loop is not formed.
Ester
The Ester category includes both ester end ether bridges, i.e. bridges characterized by the connection via C-O bond. The ester bridge arises when the free carboxylic group is condensed with hydroxyl group of threonine or serine. As the free carboxylic group is found e.g. on the C-terminus of protein, such bridge can occur upon condensation of C-terminus with the intrachain side group
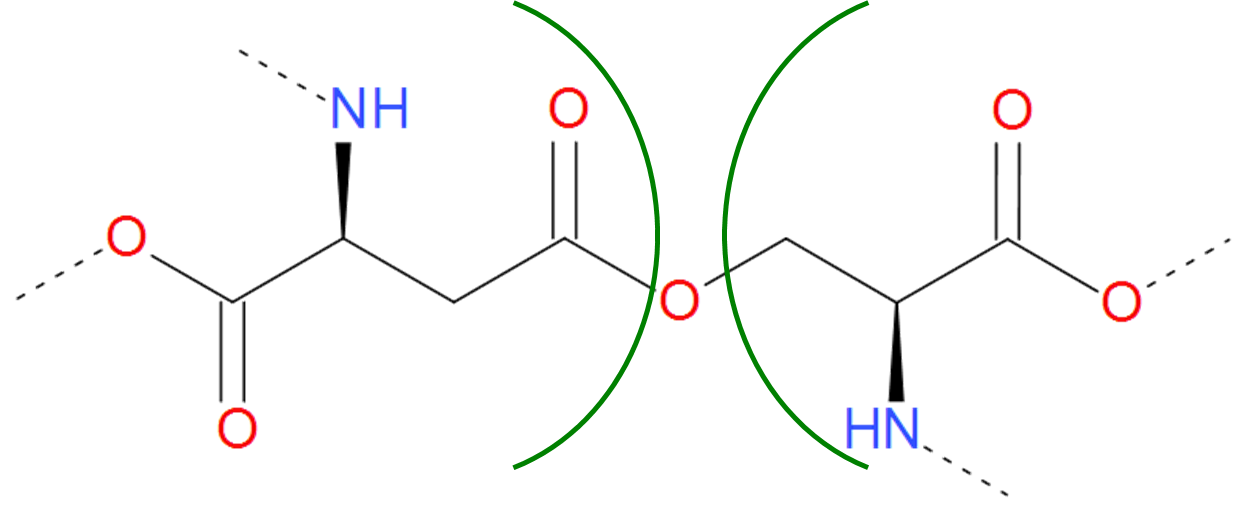
Fig. 8 Example of realization of ester bond. The residues of asparaginic acid and serine are separated by green arcs. The dashed lines denote the remaining part of the protein chain.
An ester bond is however more susceptible to hydrolysis than the amide bond. Therefore in some proteins there can be found another realization of C-O bond, namely ether bond. Ether bond is harder to be hydrolyzed, however it is also harder to be synthetized in the cell conditions.

Fig. 9 Asparagine and Valine residues joined by ether bond according to pdb file. The residues are separated by green arcs. The dashed lines denote the remaining part of the protein chain.
Thioester
The Thioester category is a sulfuric analog of the Ester category. In contains both thioester and thioether bridges, characterized by the C-S bond, which arrises in much the same way as ester and ether bridges. The only exception is, that instead of serine or threonine, one of the amino acid should be sulfur containing cysteine.
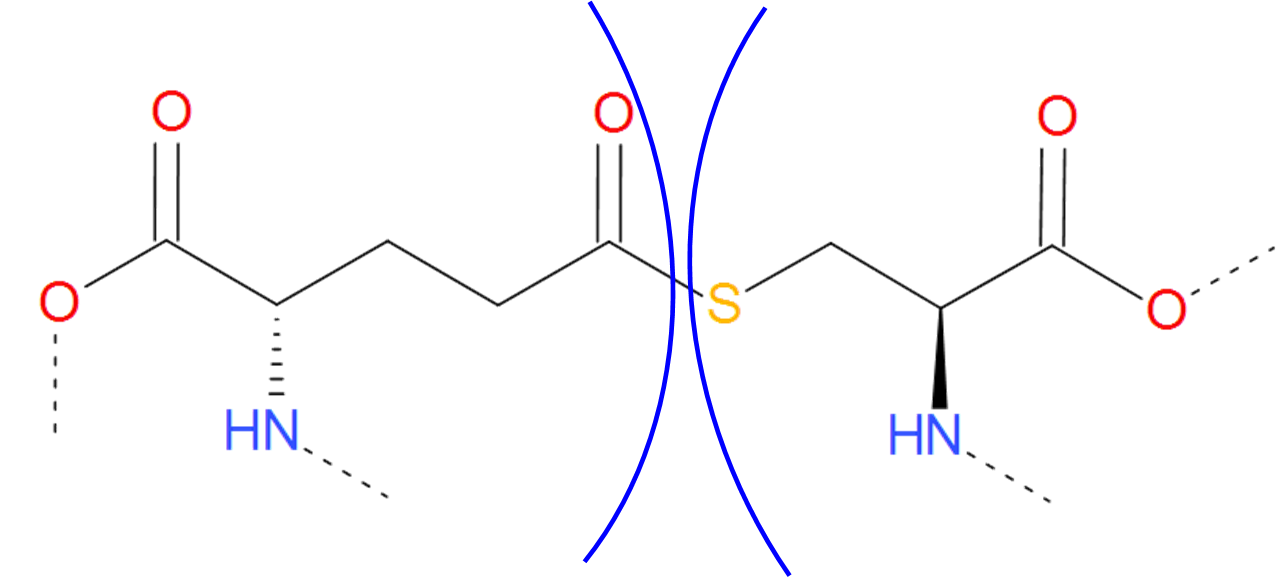
Fig. 10 Thioester bridge formed by glutaminic acid and cysteine. The residues are separated by blue arcs. The dashed lines denote the remaining part of the protein chain.
The thioester and thioether are weaker and longer, than their oxygen analogs (ester and ether). Therefore, they can be thought of as a hidden functional cysteine, which can be released in favorable conditions. Such mechanisms could be useful for example in adhesion proteins, which adhere properties would be activated in specified conditions only, and the adhering mechanism would depend on the existence of free thiol (-SH) groups.
Other
Apart from the bonds mentioned before, there are some other bridges joining side groups of proteins. These are classified together as “Other” class. The most common of them is C-C bond, often introduced artificially to increase the stability of a protein. Moreover, the C-C bond between aromatic rings of tyrosine, tryptophan, histidine or phenylalanine extends the net of conjugated bonds modifying the spectral properties of the compounds. These can be the mechanism used in e.g. photoreceptors.
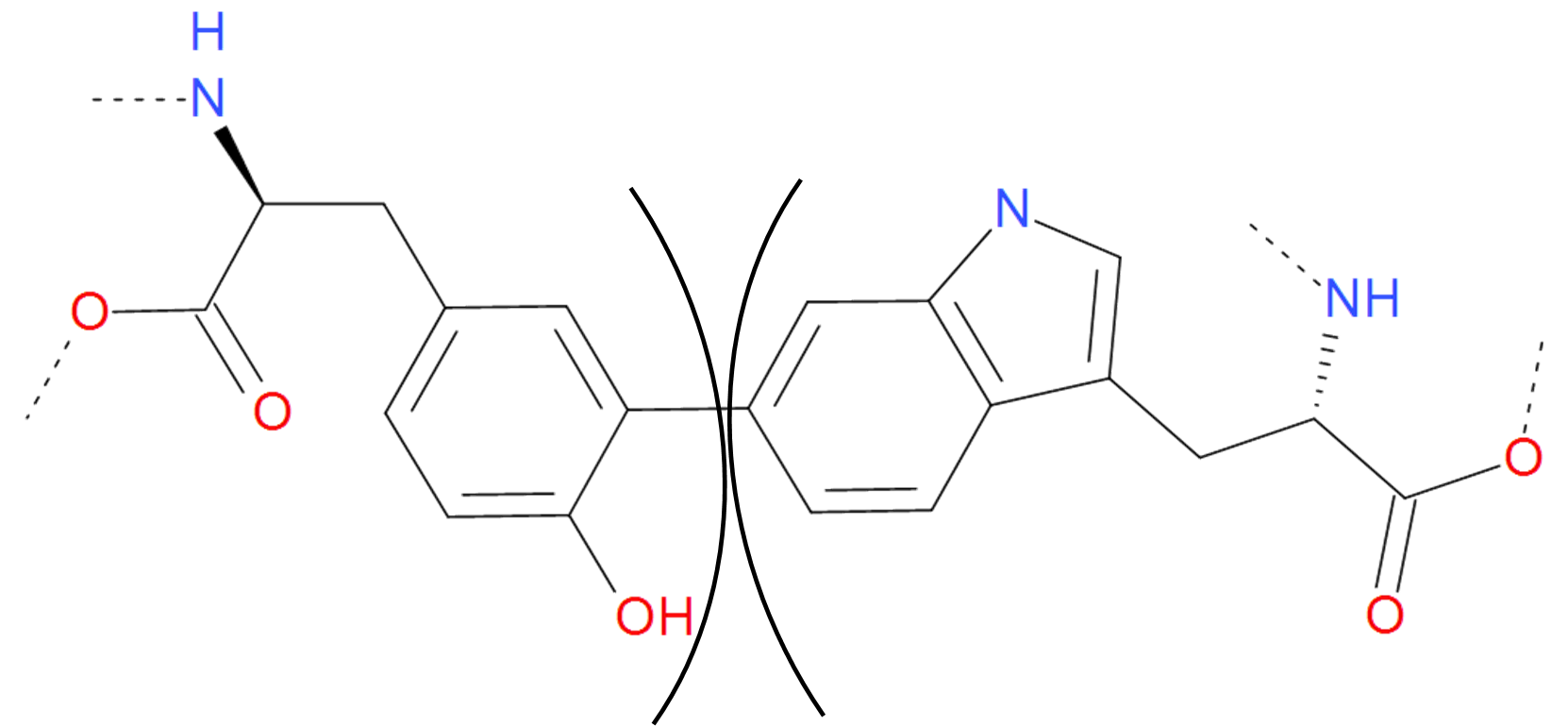
Fig. 11 The C-C bond between tyrosine and tryptophan. Two aromatic rings are joined giving rise to new structure with different spectroscopic properties. The residues are separated by black arcs. The dashed lines denote the remaaining part of the protein chain.