Search and browse database
General structure of the database
There are three main options a user can choose to view or analyze data:
- Browse database - which lists all protein structures currently deposited in the database; all these structures are also hyperlinked to other databases;
- Search database - which provides classification of proteins according to their topological, geometrical, sequential and biological properties;
- Process my structure - which allows users to upload new polymer-like structures or trajectories and analyze their lasso-type entanglement/
Apart from the above options the user can also search proteins according to their
pdb code and chain notation. These options are visible in the main page of LassoProti, as well as they are constantly visible in the top of the page (Fig. 1).
Fig. 1 The homepage of the LassoProt Server/Database with the main options in the top of the page.
Browse database
Choosing this option results in printing the list of all protein chains with non-trivial, nonartificial lasso-type entanglement deposited in the LassoProt database (Fig. 2). They can be browsed in three different ways. The default screen presents a list of proteins with their pdb codes (together with the chain number specified), identified lassos across the whole chain (see Determination of lasso types) and the title used in the pdb header. The type of chemical interaction responsible for formations of non-trivial closed loops (see Bridge determination) is indicated by an appropriate color, e.g. green indicates a loop closed by C-O bond (ester, ether) and red - a loop closed by C-N bond (amide, amine). Proteins with incomplete chains are denoted by an additional symbol of a broken chain element - .
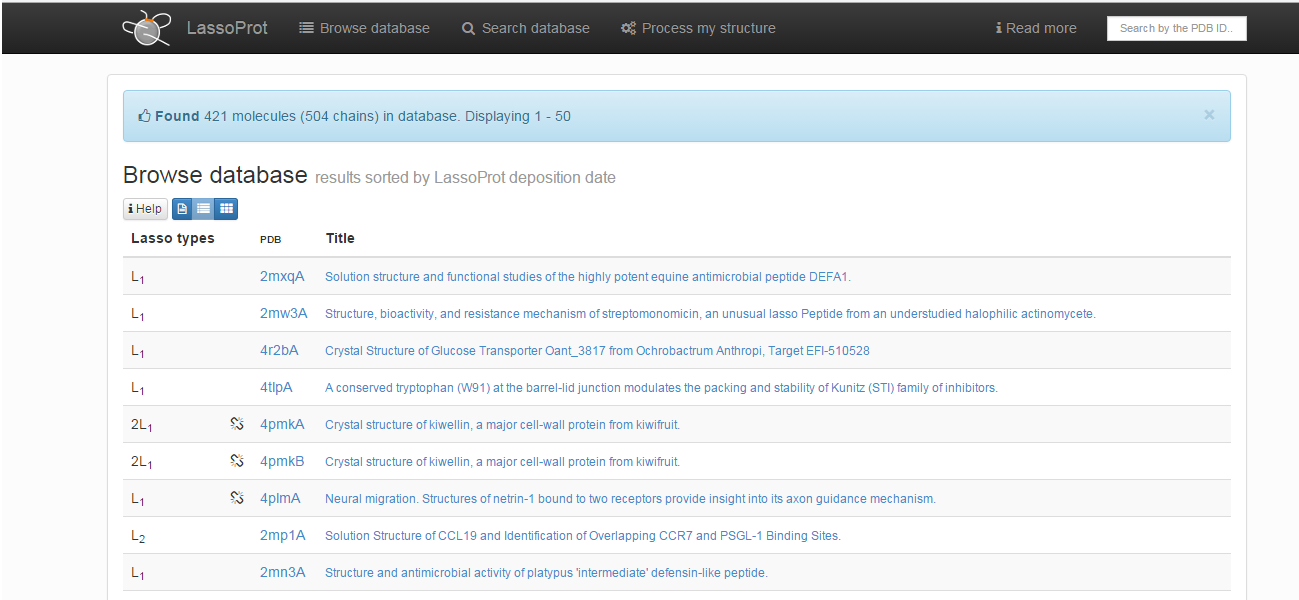
Fig. 2 The default view of the Browse section.
The next two viewing possibilities can be chosen by clicking one of the blue icons under the “Browse database” title (Fig. 3).

Fig. 3 The buttons changing the browsing options from a list of raw data (left), to a list of proteins with lasso topology and a title used in the pdb header (middle) or a set of miniature figures representing lasso entanglement as a barycentric map (right).
Fig.3 shows additional browsing options. The leftmost button enables a user to browse a list of raw data, which is suitable for independent analysis. The rightmost button enables a user to search visually through the barycentric representation (see Crossing detection or Single protein chain data presentation) of all chains (Fig. 4). Such a representation allows users to quickly identify proteins with the same lasso-type entanglemen, e.g. number and direction of crossing. However, if a protein has more than one non-trivial closed loop, only the barycentric representation for the most complicated one is presented. Such a presentation can be useful when searching for e.g. spatially close piercings. Upon choosing one of the listed proteins, the full information about it is presented as described in the section Single protein chain presentation.
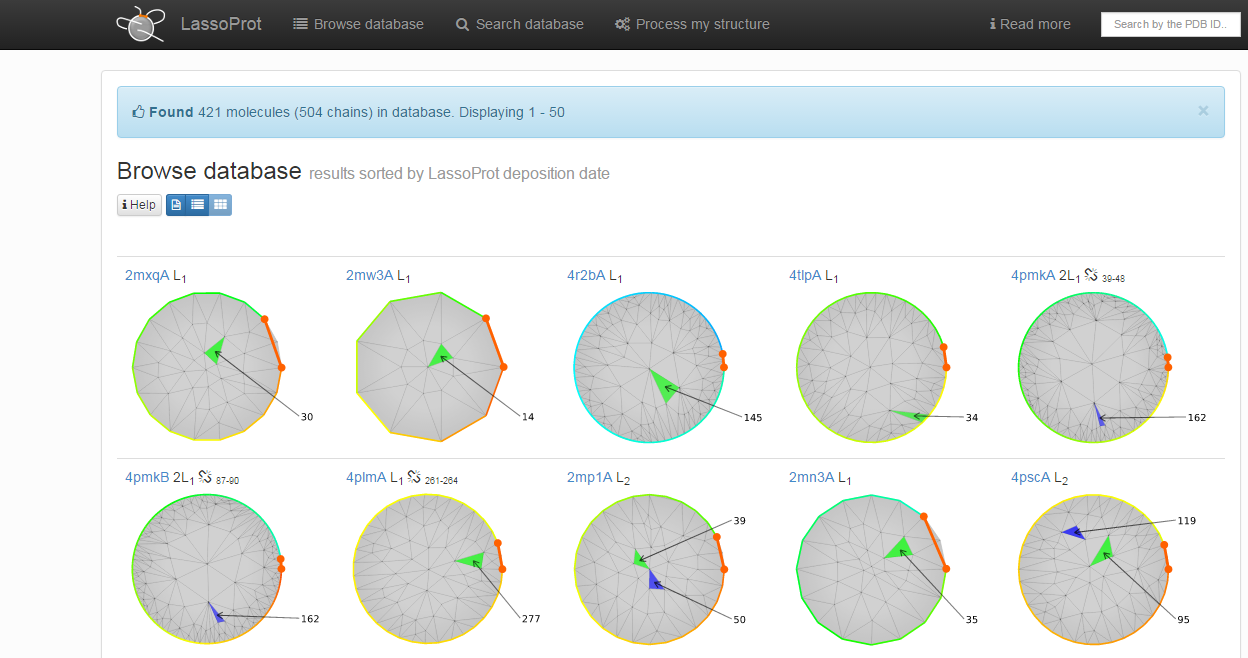
Fig. 4 Example of the view of lasso entanglement as a barycentric representation in "Browse database" mode.
Search database
A user can search the database in various ways, to find all protein chains fulfilling specific criteria. First, the user can chose between two main filters:
- Set of data: all or non-redundant set of protein chains. The non-redundant set of proteins can be useful when analyzing the statistics of class cardinality, loop/tail lengths or the loop area.
- Type of bridge: SS-bridge, Amide, Ester, Thioester, Other. This filer enables the user to analyze only the structures with the chosen bond closing the loop (see Determination of bridge type).
Both these filters are set for every tab (Loops, Molecule keywords etc.) described in detail below, during the search.
The default screen is the Lasso type screen in which the following options can be chosen (Fig. 4):
- “Lasso types”: enables the user to print structures containing covalent loops with defined lasso type. More than one type of lasso-type entanglement can be chosen from five major classes (L1, L2, L3, LS, LL). After choosing a major topological class, the user can define a subclass of interest that includes some specific geometrical information: the tail which pierces the surfaces (N- or C-terminal) and the direction of piercing the surface. After choosing an appropriate group to be shown, the user can decide if broken structures (structures with missing coordinates of some Cα atoms) should be printed. Apart from major classes the user can also search through L0 class in which the proteins with unpierced closed loop (trivial loops) are stored, and the Artifact class. In the latter one the structures with a gap larger than 6 residues and those to which correctness we had doubt (improper distance between Cα atoms) are stored. For chains in the Artifact class the topology can be non-trivial, however one has to be very carefull with interpreting the results, as the topology can be a result of e.g. errors in crystallization.
- “Fingerprint”: shows the set of all types of identified lassos along one protein chain (starting from the N-terminus) with cardinality of each topological class, such as L1L3, LS22L1LS2. For each fingerprint a user can print structures with such a lasso pattern by clicking on a symbol.
- “Lasso loop length”, “Lasso tail lengths”, “Lasso Loop area”: chains are grouped according to the length of the closed loop and tails, and the closed loop areas, and shown as a histogram of occurrences of each value. By clicking on the definite histogram box a user can print all structures with a chosen loop/tail length or the closed loop area.
Apart from the main panel (“Lasso types”) the user can check the entanglement motif of structures with a chosen Molecule keyword, Molecule tag (based on the classification from the
pdb website),
pfam family identifier,
ec nomenclature (numerical classification for enzymes based on the chemical reactions they catalyze) or
cath classification (which includes class, architecture, and topology). Moreover one can get a gist of the proportion between the function of different complex lasso proteins by choosing “Keywords cloud” option. We believe that based on these classifications, the database will be easily searchable and will easily fit to the needs of all users, providing the opportunity for other researchers to make new discoveries.
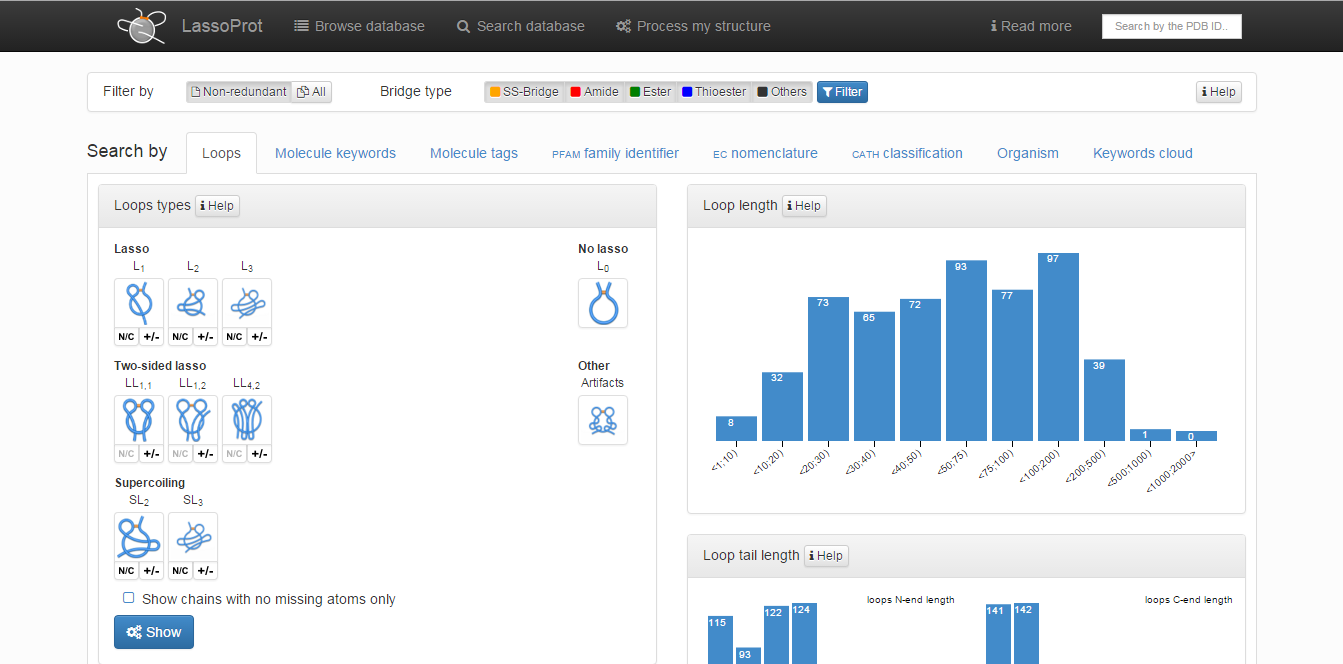
Fig. 5 The main view of the search tab.
Process my structure
Users can upload and analyze two types of data: either a single structure, or the whole set of structures (e.g. a folding or unfolding trajectory of a protein). The data can be submitted either in pdb format or in a simplified xyz format (containing only indices and cartesian coordinates of atoms, which enables users to analyze arbitrary polymers). Moreover, users can choose between:
- automatic detection of five types of chemical interactions responsible for formation of closed loop (S-S, N-O, C-O, C-S and other);
- choose one of the loop-closing chemical bond;
- define the position of the bridging residues.
Additionally via the
Advanced options users can establish new geometrical conditions to detect piercing through a minimal surface. Those extended options allow users to adjust this tool to their needs and to monitor any surfaces they wish. In case of a trajectory, an
xtc format (typical for Gromacs software) can also be uploaded (with a
gro or
pdb file of starting frame).
After analysis in the single structure case, except of topological information, the user can download necessary files to visualize the structure along with the determined surface in
vmd and
mathematica. Additionally users can download smooth representations of lasso proteins (in
pdb format), which is very helpful in understanding lasso topology. The full description of how to process structures uploaded by users is described in
Server - Analyze single structure and trajectory section.