Proper analysis of complex lasso proteins and (bio)polymers requires investigation of its dynamics. This, on the other hand requires the ability to study in detail also a selected frame. LassoProt provides both options to the users.
In both cases (single frame as well as trajectory analysis mode) the LassoProt server supports few basic formats of input files and gives the user a set of advanced options that allow to adjust this tool to the users needs. Additionally, the files enabling visualization of the structure with the surface spanned on the closed loop can be downloaded. Currently LassoProt provides files for vmd and Mathematica. Such presentation would certainly help the users to imagine the notion and location of minimal surface spanned on the closed loop and can be essential in further personal analysis. In case of trajectory, the server displays dynamic plots presenting the change of lasso type during the dynamics, with Rgyr plot for comparison.
To further facilitate the understanding of the lasso-type entanglement, in single frame analysis mode smooth representation of lasso proteins or (bio)polymer (in pdb format) is available as well. All the details concerning single structure, or trajectory are described in the following subsections:
Input
Coordinates of the structure can be provided in three ways:
1 -9.102 -18.555 15.000 2 -9.384 -17.556 14.080 3 -5.661 -16.841 14.367 4 -3.660 -15.387 11.487 5 0.096 -15.769 11.241 6 2.646 -13.739 9.329 7 5.806 -15.775 8.604 ...
Remark: In pdb file all information should be located in their proper position in line. In particular, the "CA" atom name should be placed in 14th and 15th characters of line. This notation (although not required exactly by pdb standard) is widely used. Please note however, that some format converting software shift the "CA" characters. E.g. CatDCD locates "CA" in 15th and 16th character of the line. Please note also, that the chain name is required to be present in the pdb file.
After selecting the file, the user is supposed to choose the covalent loop (or the loops - as many as wanted), which ought to be analyzed. In case of original pdb file (wiht header) this can always be done in three ways (Fig. 1).
The loop closing bond can be either disulfide (S-S), amide (or amine, N-C), ester (or ether, C-O) thioester (or thioether, C-S) or other. More information about possible loop closing bonds with examples are given in bridge type determination section. The automatic detection of the loop closing bonds is possible only in the original pdb files containing appropriate information in SSBOND or LINK lines.
The uploaded file is checked to confirm that it contains proper protein data. The conditions which protein chain must fulfill are basically the same, as in the case of the data stored in the database (see Closed loop detection). Those are:
Unticking this option gives user additional possibilities to establish new geometrical conditions to distinguish between artificial (uncertain) and correct (stable) piercings - setting the minimal amount of residues between two piercings, between piercing and a closed loop and between piercing and closer end of a chain (see details in Reduction of artificial piercings section).
To restore the default settings the user can simply tick "Stable lasso" option.
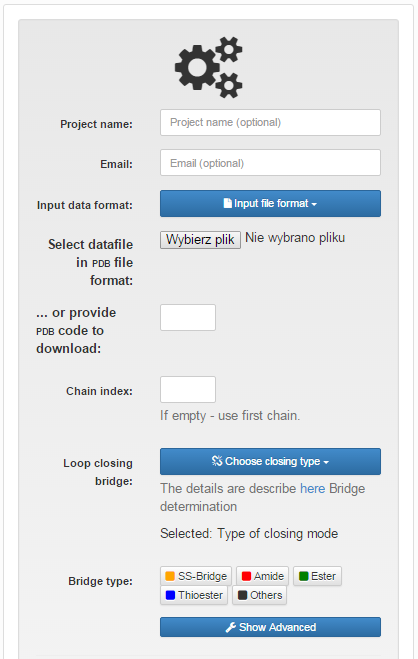
Fig.1 The view of the input page for single structure with "Choose type of closing loop" option selected.
Output
The results of the single structure analysis are presented in the same way as the information about any chain in the LassoProt database, described in details in the Single protein chain data presentation section (Fig. 2).
In particular, the user can download all information about a lasso entanglement and visualization facilitating interpretation of the data. In particular users can download the structure in pdb and xyz format, files for vmd, Mathematica and JSmol, enabling them to view the minimal surface spanned on the closed loop, and finally the image with barycentric representation of the surface, in svg format or as Mathematica script. The file for vmd is written as a tcl script. To view the surface, one has to first open the structure in vmd (the downloaded pdb file), click Extensions-> TK Console and load the tcl script in the new window via File->Load file.
Additionally, user can download all files with smooth representation of the structure. This could be helpful in understanding lasso-type entanglement in more complicated proteins. The LassoProt uses the procedure where chain is being smoothed via averaging coordinates of three neighbouring atoms, as long as the lasso type of the structure stays unchanged (the running average), or 16 runs are done (the sufficient number in all known cases). The number occuring at the end of the “smooth files” names indicates the number of iterations of that procedure that were applied to a chain.
During the analysis there some errors concerning the input file may occure. We provide a full list of known problems with possible solutions in the Possible errors subsection.
A structure uploaded and analyzed by the user is stored for 14 days (so that it can be viewed again).
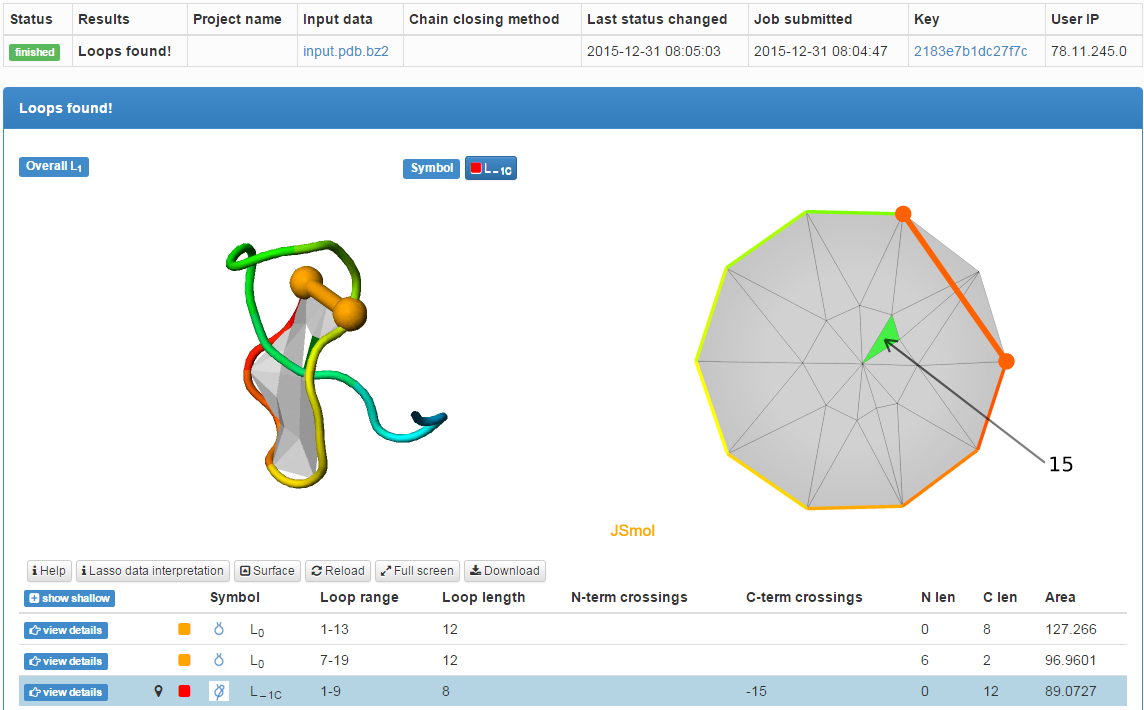
Fig. 2 The typical output for a single structure analysis. Two disulfide bridges and one amide bridge were detected. The surface spanned on the loop closed by amide bond is pierced once.
Input
A trajectory can be uploaded in three different formats:
t 0 1 -9.102 -18.555 15.000 2 -9.384 -17.556 14.080 3 -5.661 -16.841 14.367 4 -3.660 -15.387 11.487 5 0.096 -15.769 11.241 6 2.646 -13.739 9.329 7 5.806 -15.775 8.604 t 1 1 -9.102 -18.555 15.000 2 -9.384 -17.556 14.080 3 -5.661 -16.841 14.367 4 -3.660 -15.387 11.487 5 0.096 -15.769 11.241 6 2.646 -13.7399.329 7 5.806 -15.775 8.604 t 2 ....
Remarks: The pdb format requires correct positioning of information in each line. In particular, the "CA" name should appear as 14th and 15th character of the line (this concerns especially CatDCD users, as this software moves the "CA" name one field to the right). Moreover, the chain name is required in each frame. Also please pay attention to separation of models in pdb file. The models should be separated by both "ENDMDL" and "MODEL n" lines (where "n" is the number of the frame). The models separated by "END" lines are not valid. If any doubts please compare your file to Exemplary input files.
In the case of trajectory a user is always prompted to determine only one bridge by entering bridge forming residues indices (we use the notation from pdb file and we do not renumber the indices).
In each case the user can determine the level of calculation complexity, and level of output complexity ("More detailed algorithm" and "More detailed output file"). Finally, user can define the "Step" i.e. number of frames which should be skipped when analyzing the trajectory. Increasing this value speeds up the calculation (default value is 1).
In advanced options, the user can also define own criteria of structure treatement. For that end, the user should untick the "Stable lasso" option. Than four parameters can be changed (Fig. 3):
Remark Note, due to algorithm used, the structure uploaded would still rise an error if consecutive atoms were too close to each other (less than 0.1 Å) notwithstanding unticking "Ignore bad length..." option.
If the files are correct, the user is notified by the communique on the page. Finally the analysis of the topological state of a chosen covalent loop, the piercings list and the surface area are being counted.
Remark: The maximal size of uploaded trajectory is 20mb.
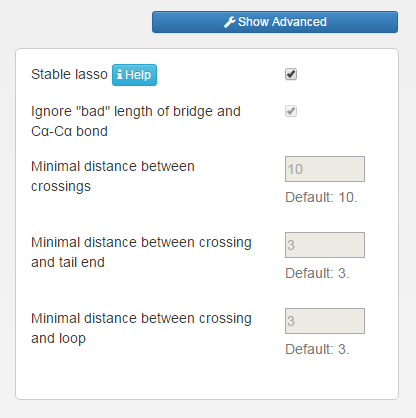
Fig.3 The view of advanced options in trajectory mode.
Output
The output for trajectory mode is suited for the analysis of the lasso entanglement dynamics. First, we provide two charts: a) one displaying the change of lasso type in consecutive frames; b) showing the indices of piercing residues (Fig. 4). Both plots are dynamical i.e. additional information are shown upon pointing the mouse above chosen frame. To facilitate analysis the charts can be also zoomed by selecting region of interest by cursor. The change in lasso type and the piercing residue index can serve as new reaction coordinate (see Apply results section). Therefore we compare it with standard reaction coordinate Rgyr shown as a orange subplot in the second chart. This option is avaible only upon choosing "More detailed output" in the previous step.
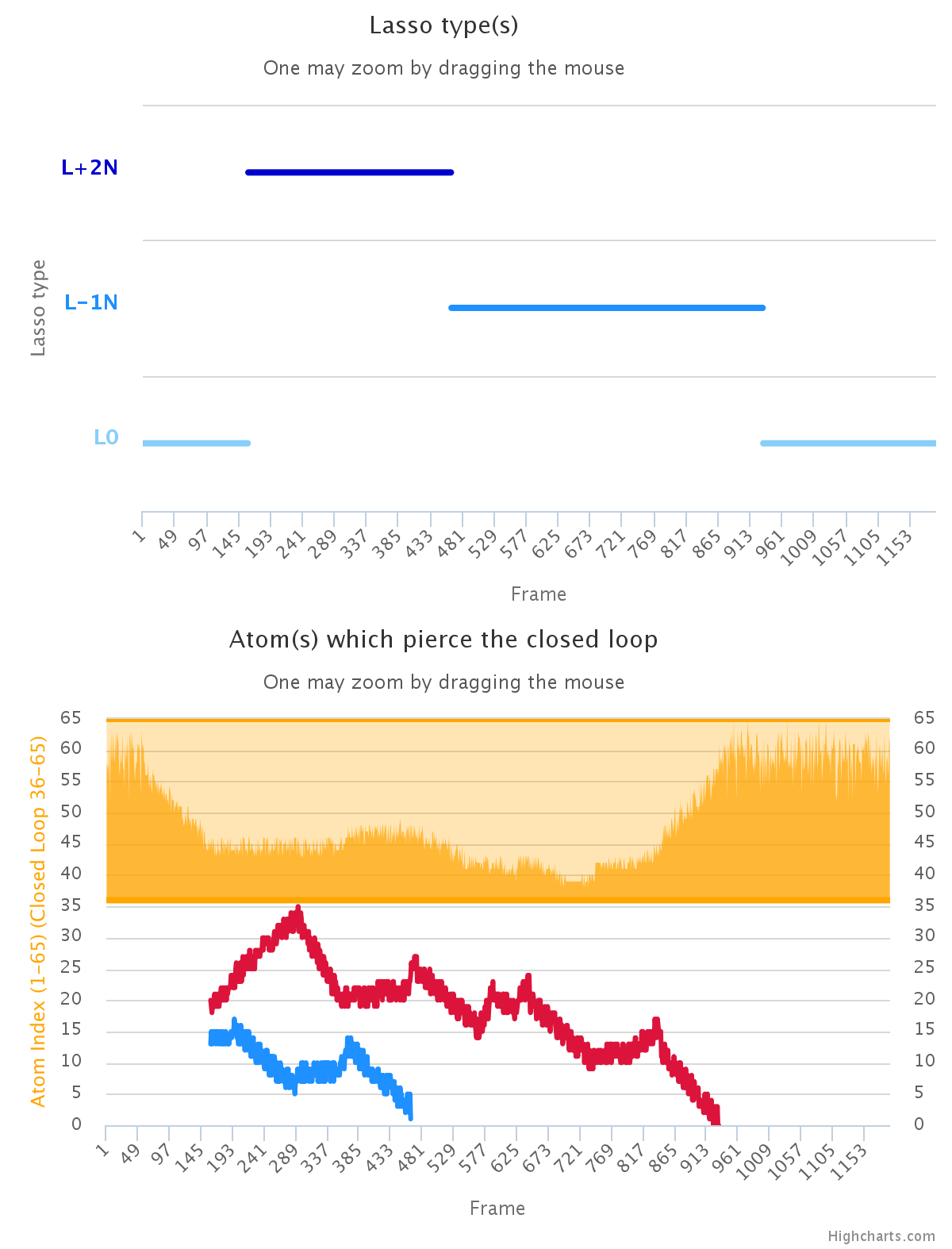
Fig. 4 The exeplary charts showing the dynamics of lasso type (upper plot) and the index of piercing residue (lower panel). For comparison, the orange sublot in lower panel shows Rgyr for analysed system.
Below the plots, in the grey box the raw data are shown. The content of the box differs depending on the compelexity of the output chosen. In both cases the user can download the result as separate text file for further analysis. The example of less detailed output file is given below:
#Path to the analyzed file: example.xyz #Id_begin_chain id_end_chain id_begin_loop id_end_loop: 1 65 36 65 #More detailed algorithm: False #More detailed output: True #Checked distances in the chain: True #Minimal distance between crossings (not to be reduced): 10 #Minimal distance between crossing and tail end (not to be reduced): 3 #Minimal distance between crossing and loop (not to be reduced): 3 1 1 0 | -26 | | L-1N 2 1 0 | -25 | | L-1N 3 1 0 | -26 | | L-1N ...
In the first 8 lines (with # character) the description of the file content is shown. The first line contains the name of analyzed file. Second contains the indices of the N- and C-terminal residues and the indices of bridge-forming residues. Below are the lines summarizing the option chosen.
The following lines contain the lasso data for each frame of trajectory. The number in the first column is the number of the frame in the trajectory. The next two numbers describe how many piercings there are formed by N- and by C-terminus respectively, after reduction of shallow (artificial) piercings (see details in Reduction of artificial piercings section). In the center, between # signs a list of piercings is presented, where N- and C-terminal crossings are separated by the central # sign. The last column shows the lasso type determined for a given closed loop.
The example of more detailed output file, contains more information:
#Path to the analyzed file: example_more_detailed.xyz #Id_begin_chain id_end_chain id_begin_loop id_end_loop: 1 65 36 65 #More detailed algorithm: False #More detailed output: True #Checked distances in the chain: True #Minimal distance between crossings (not to be reduced): 10 #Minimal distance between crossing and tail end (not to be reduced): 3 #Minimal distance between crossing and loop (not to be reduced): 3 1 1 0 1 X -26 X XX 1 0 X -26 X XX L-1N XX 22.7195 2 1 0 1 X -25 X XX 1 0 X -25 X XX L-1N XX 20.6149 3 1 0 1 X -26 X XX 1 0 X -26 X XX L-1N XX 20.3387 4 1 0 1 X -25 X XX 1 0 X -25 X XX L-1N XX 22.1802 ...
As previously, the first 8 lines are description of file content. Then, the number in the first column is the frame number and the next two values describe how many piercings are there formed by N- and by C-terminus respectively, but in this case before any reduction. In the fourth column there is an information about surface orientation (it is possible for some surfaces to be unorientable, a minimal surface spanned on Möbius strip is the example; however, in the case of proteins we have not found such an example). After the X sign there is a list of piercings before any reduction, where N- and C-terminal piercings are separated by the next X sign. Then, after XX sign there is an information about lasso type after piercing reduction. In particular two following values describe the number of N- and C-terminal piercings (after reduction). Then, after the next X sign there is a list of those „deep” piercings, where N- and C-terminal crossings are separated by next X sign. The last columns contain the lasso type determined for a given covalent loop, and radius of gyration separated by XX sign.
Remark: The time of calculation strongly depends on the number of frames in the trajectory. Although the maximum size of uploaded file is 20mb, we strongly recomend uploading files with small number of frames, for example up to 2000 frames. For longer trajectories the analysis is expected to significanlty more time. E.g. the 14 000 frames trajectory is analyzed about 5h.
The trajectory analysis is compatibile with single frame mode. Below the trajectory data, an exemplary frame is presented as in single structure mode. Even lower the user can specify, which frame should also be analyzed in detail, in single frame analysis mode. The results of such analysis are shown in a separate window.
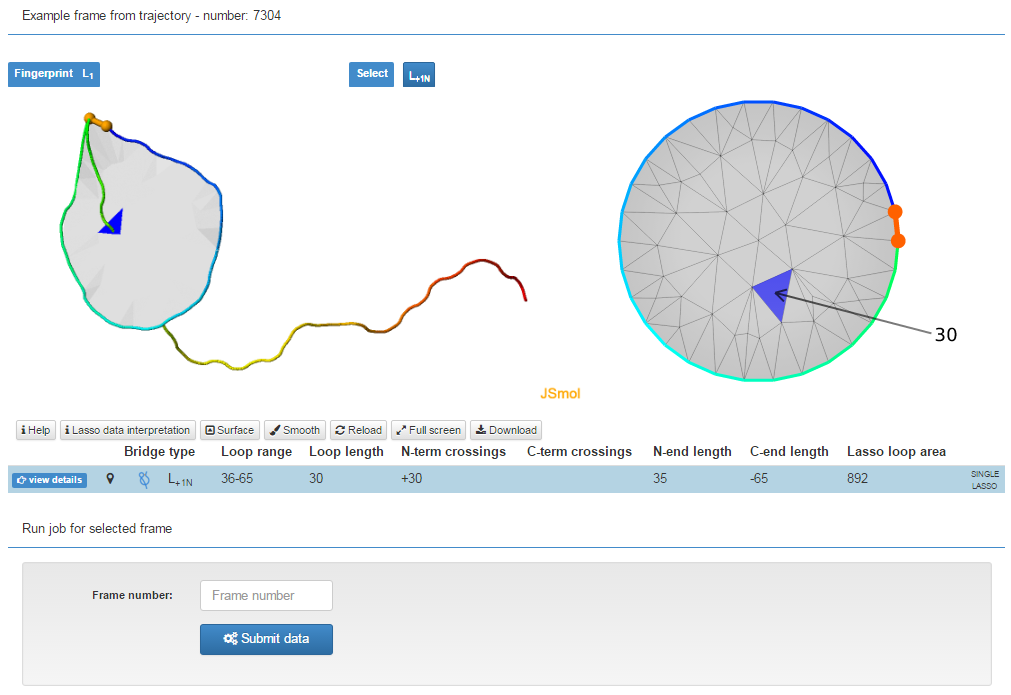
Fig. 5 The exeplary trajectory frame with the box in which the user can specify, which frame should be analyzed.
Remark: Only frame numbers which exactly match the values in the trajectory output (grey box) will be accepted.
As in case of single frame, the result of trajectory analysis are stored for 14 days.
During the analysis some errors concerning input file may occure. We provide a full list of known problems. Clicking on them will display possible solutions. If after reading all known solutions you are sure, that the file is correct and contains the Cα coordinates written in proper way, please send us the file, as that could be a clue how to fix and improve our server.